2.4. The p53 pathway
2.4.1. TP53 gene and p53 protein
The tumour suppressor protein p53 was first described in 1979 (Lane & Crawford 1979, DeLeo et al. 1979, Linzer & Levine 1979) and ten years later identified as a tumour suppressor (reviewed by Levine 1990). In human, the TP53 gene that contains 11 exons is located in chromosome 17p13.1, while the mouse gene, which also contains 11 exons, is located in chromosome 11 (reviewed by Soussi & May 1996). In both human and mouse, the coded protein is approximately 53 kDa in size, the human protein containing 393 amino acids and mouse protein 390 amino acids. The p53 protein consists of an acidic N-terminus with a transactivation domain, a hydrophobic central DNA-binding core and a basic C-terminus with regulatory and oligomerisation domains (for a review, see e.g. Hainaut & Vähäkangas 1997). The DNA-binding domain of the p53 protein is composed of two beta-sheets and a zinc atom which stabilizes the structure (Cho et al. 1994).
2.4.1.1. Functions of the p53 protein
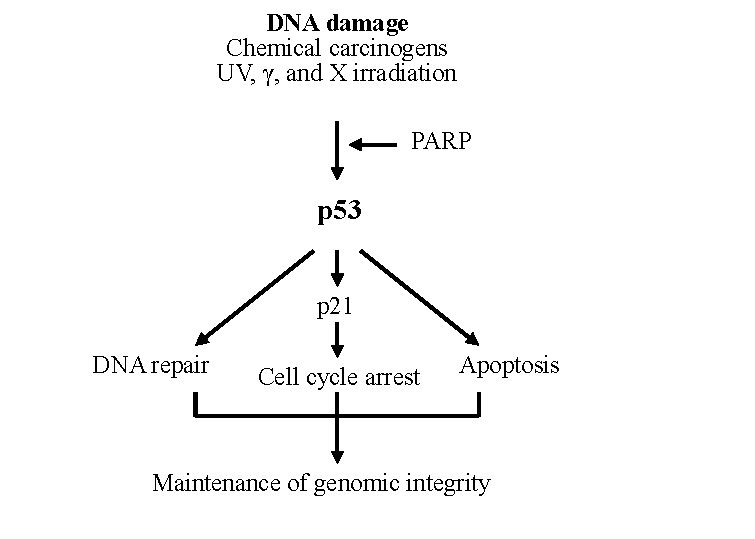
It is nowadays known that p53 is not functional or functions incorrectly in most human cancers, and that it plays a crucial role in the prevention of tumour development (reviewed by Vogelstein et al. 2000). The general assumption is that the p53 network in normal, non-activated situations is non-functional, but is activated in cells as a response to various signals that take place in the carcinogenic process (for a review, see Vogelstein et al. 2000). Carcinogen-induced DNA damage (Fig. 2), abnormal proliferative signals, hypoxia and loss of cell adhesion are some of the most common signals. It is thus likely that p53 takes part in several stages of the carcinogenic process (for a review, see Woods & Vousden 2001). The importance of a functional p53 protein is emphasized by the fact that p53-deficient mice show a very high incidence of multiple, spontaneous tumours at an early age (Donehower et al. 1992, Donehower et al. 1995).
Of the many functions of p53, the first ones identified were inhibition of abnormal growth of cells (reviewed by Sionov & Haupt 1999) and triggering of programmed cell death (Fig. 2, Heinrichs & Deppert 2003). Because these processes ensure genomic integrity or destroy the damaged cell, p53 has been called the “guardian of the genome” (Lane 1992). Later on, other important functions, such as DNA repair (reviewed by Albrechtsen et al. 1999) and inhibition of angiogenesis (for a review, see Vogelstein et al. 2000), were discovered. p53 is a sequence-specific nuclear transcription factor that binds to defined consensus sites within DNA as a tetramer and affects the transcription of its target genes (El-Deiry 1998). p53 regulates these genes either by transcriptional activation (Murphy et al. 1999) or by modulating other protein activities by direct binding (Guimaraes & Hainaut 2002). The p53-induced activation of target genes may result in the induction of growth arrest either before DNA replication in the G1 phase of the cell cycle or before mitosis in the G2 phase. The growth arrest enables the repair of damaged DNA. By programmed cell death, which is often referred to as apoptosis according to its morphological appearance, the cells damaged beyond repair are eliminated thus preventing the fixation of DNA damage as mutations. An interesting question is what determines the choice between growth arrest and apoptosis. Extensive studies on this topic have been made, and several factors influencing the response of the cell have been suggested: cell type, oncogenic cell composition, intensity of stress, level of TP53 expression, interaction of p53 with specific proteins and affinity of p53 for promoters (reviewed by Vousden & Lu 2002). However, the details of how p53 prevents the accumulation of mutations after DNA damage still require further studies.
2.4.1.2. Regulation of p53
The p53 protein is effectively able to inhibit cell growth, and its activity is therefore strictly regulated. There are several mechanisms for the regulation of p53. Although, in some models, chemical DNA damage, by BP, for example, seems to increase TP53 transcription (Pei et al. 1999, Lu et al. 2000), it is generally believed that the principal mechanisms governing the activity of p53 occur at the protein level. These include post-translational modifications, regulation of the stability of p53 protein, and control of its sub-cellular localization (for a review, see Woods & Vousden 2001). Post-translational modifications of the protein take place in response to stress, and different agents elicit diverse responses (reviewed by Appella & Anderson 2001). The human p53 protein has been shown to be modified at least at 17 different sites (for a review, see Appella & Anderson 2000). Of the post-translational modifications of p53, the most widely studied and best-known so far is phosphorylation. After DNA damage induced by ionizing radiation or UV light, phosphorylation takes place mostly at the N-terminal domain of p53 (reviewed by Appella & Anderson 2001). Another important modification is acetylation, which has been shown to occur in response to chemically induced DNA damage and hypoxia (Ito et al. 2001). In response to DNA damage, the p53 protein is also modified by conjugation to SUMO-1, a ubiquitin-like protein (Gostissa et al. 1999). Many proteins able to interact with p53 may also play a role in p53 regulation (reviewed by Vousden & Lu 2002).
Mdm2-mediated degradation regulates the stability of p53. The relationship between mdm2 and p53 is discussed in more detail in chapter 2.4.2. For many of its functions, p53 needs to be localized in the nucleus. The p53 protein involves nuclear import and export sequences, and the activity of p53 is regulated by both nuclear import and nuclear export (Stommel et al. 1999, for a review, see Vousden & Vande Woude 2000). The p53 protein needs an ability to interact with microtubules in order to move close to the nuclear import machinery in the cytoplasm (Giannakakou et al. 2000). Also, any changes in the conformation of the p53 protein and the regulation of its DNA-binding activity affect its function (Hupp et al. 1992, Cain et al. 2000).
2.4.1.3. Aberrations of p53 function
There are many ways in which the p53 function may be altered in human cancers. p53 can be inactivated indirectly through binding to viral proteins, as a result of alterations in the mdm2 or p19ARF genes or by localization of the p53 protein to the cytoplasm (for a review, see Vogelstein et al. 2000). The most common aberration of p53 in human cancers is, however, mutation of the TP53 gene. A database of TP53 mutations is maintained by IARC (http://www.iarc.fr/p53/). Most of the mutations in the TP53 gene occur in the exons 4-9, the coding region for the DNA-binding central domain of the protein. A large proportion of all mutations in TP53 are single base substitutions (87%, Hainaut & Hollstein 2000). Of all mutations, approximately 30% occur in six codons (175, 245, 248, 249, 273 and 282, Greenblatt et al. 1994), which are called the hotspot codons. These residues are located in the DNA-binding part of the protein (Cho et al. 1994), and mutations in these codons influence the protein-DNA contacts and the conformation of the protein (reviewed by Guimaraes & Hainaut 2002). It also seems that, in cancer cells with normal TP53 alleles, the expression or regulation of the protein is often somehow altered. Other factors that prevent normal folding of the p53 protein, such as cadmium, may influence its DNA-binding capacity (Méplan et al. 1999). It has therefore been suggested that all cancer cells have some aberration of p53 (Guimaraes & Hainaut 2002).
2.4.2. Mdm2 protein
The murine double minute 2 (mdm2, hdm2 in human) gene encodes a 90 kDa protein (97 kDa in human) that was originally identified as a dominant transforming oncogene (Fakharzadeh et al. 1991). The mdm2 gene has been found to be amplified in human cancers (reviewed by Momand et al. 1998). The combination of overexpressed mdm2 and p53 gives a worse prognosis than either one of them alone (Würl et al. 1998). Deletion of the mdm2 gene in mice is embryonically lethal, probably due to increased accumulation of p53, but this lethality can be counter-acted by deletion of the TP53 gene (Jones et al. 1995, Montes de Oca Luna et al. 1995). The p53-mdm2 relationship (see Fig. 3) is vital in the regulation of cell growth and death.
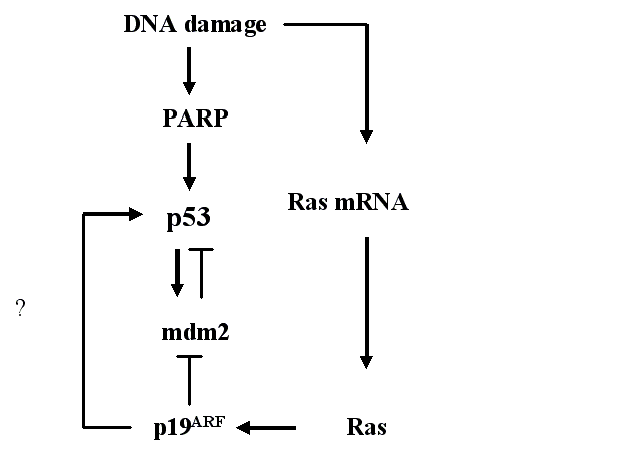
The mdm2 protein regulates the activity of the p53 protein with more than one mechanism. It can block the transcriptional activity of the p53 protein, export p53 from the nucleus to the cytoplasm and promote the degradation of p53 (for a review, see Alarcon-Vargas & Ronai 2002). The mdm2 protein functions as a ubiquitin ligase and can ubiquitinate p53 (Honda et al. 1997, Fuchs et al. 1998). Ubiquitination targets the p53 protein for degradation by the proteasome. Post-translational sumoylation of mdm2 increases this ubiquitination, whereas DNA damage reduces the sumoylation of mdm2 and thus the ubiquitination of p53. Hence, the amount of p53 increases (Buschmann et al. 2000, Buschmann et al. 2001). p53, on the other hand, affects the amount of mdm2 by positive transcriptional activation, creating a feedback loop (for a review, see e.g. Oren et al. 2002).
The mdm2 protein is able to shuttle between the nucleus and the cytoplasm (Roth et al. 1998), and it is known to bind to the p53 protein in the N-terminal region (Chen et al. 1993, Picksley et al. 1994). Through binding to p53, mdm2 shuttles p53 out of the nucleus to the cytoplasm for degradation (Freedman & Levine 1998). DNA damage induces phosphorylation of the p53 protein at multiple sites, including those overlapping with the mdm2-binding sites, thus preventing the association between mdm2 and p53 (Shieh et al. 1997).
2.4.3. p19ARF protein
The gene that encodes the p16INK4a protein also encodes p19ARF (ARF, Alternative Reading Frame). However, the p19ARF protein is expressed by a separate promoter with a different first exon (1, Mao et al. 1995, Duro et al. 1995). Both p16INK4a and p19ARF function as tumour suppressors (for a review, see Zhang & Xiong 2001). Selective deletion of the p19ARF exon 1 in mice results in the development of spontaneous tumours at an early age (Kamijo et al. 1997). In cells, the p19ARF protein normally localizes to the nucleolus (Weber et al. 1999). p19ARF is able to bind the central or C-terminal portion of the mdm2 protein (Zhang et al. 1998).
The p19ARF protein is capable of interacting with mdm2 (Kamijo et al. 1998) and interfering with the autoregulatory feedback loop between the p53 and mdm2 proteins (Fig. 3), thus increasing the amount of p53. There are currently three competing theories about how p19ARF inhibits mdm2-mediated p53 degradation. The first possibility is that the p19ARF protein sequesters the mdm2 protein into the nucleolus, thus releasing p53 (Tao & Levine 1999, Weber et al. 1999). The second model suggests that nucleolar p19ARF is relocalized by mdm2 to the nucleoplasm and forms a ternary complex with mdm2 and p53, thus blocking the nuclear export of both mdm2 and p53 (Zhang et al. 1998). p19ARF has also been shown to bind the p53 protein directly, indicating that it can, in addition to mdm2, recruit p53 into ternary complexes (Kamijo et al. 1998). The third model proposes that, because the p19ARF protein is able to bind to the mdm2 protein and inhibit its ubiquitin ligase activity, p19ARF might prevent p53 nuclear export by blocking the ubiquitination of p53 (Honda & Yasuda 1999). It was shown by Weber and coworkers (2000) that triple knock-out mice lacking functional p53, mdm2 and p19ARF proteins develop tumours at a greater frequency than mice lacking p53 and mdm2 or p53 alone. This suggests that p19ARF is a tumour suppressor independent of mdm2 and p53. The p19ARF protein itself is regulated primarily at the transcriptional level. Both Myc and E1A oncoproteins have been shown to induce the synthesis of p19ARF (Zindy et al. 1998, De Stanchina et al. 1998, respectively). In summary, these three proteins form a system that regulates their localization, amount and function (Fig. 3).
2.4.4. Other p53-related proteins
2.4.4.1. Poly(ADP-ribose)polymerase
Poly(ADP-ribose)polymerase (PARP) has long been known to play a role in the recognition of DNA damage and in DNA repair (for a review, see Tong et al. 2001). One of the first events to take place after DNA strand breakage, caused either directly by genotoxic agents or indirectly by enzymatic incision of a DNA-base lesion, is the increased synthesis of poly(ADP-ribose) by PARP (Fig. 3). This is followed by poly(ADP-ribosyl)ation of proteins localized near the DNA strand breaks. The PARP protein detects DNA strand breaks and catalyzes the attachment of ADP-ribose units from NAD to itself and to other proteins (reviewed by Lindahl et al. 1995). The substrates of PARP can then influenze the architecture of chromatin or act in DNA metabolism.
PARP is known to be involved in the regulation of p53, although the relationship is not clear. There are studies showing that PARP upregulates p53. Cell lines derived from Chinese hamster V79 cells that are defective in poly(ADP-ribosyl)ation have lower basal p53 levels and fail to activate p53 in response to etoposide (Whitacre et al. 1995). Also, in lymphoblastoid cells with normal PARP activity, the PARP inhibitor 3-aminobenzamide (3AB, Purnell & Whish 1980) suppresses the accumulation of p53 after BPDE (Venkatachalam et al. 1997). On the other hand, contradictory results have also been published. X-ray-induced p53 accumulation was prolonged in mouse prostate cells treated with a PARP inhibitor 3AB (Lu & Lane 1993). Also, alkylating N-methyl-N-nitrosourea (MNU) caused increased accumulation of p53 in PARP-deficient splenocytes (Ménissier de Murcia et al. 1997). It has been suggested that PARP plays a positive role in the activation and upregulation of p53 (Malanga et al. 1998). Smulson and coworkers (2000) have shown that, in human osteosarcoma cells, p53 is poly(ADP-ribosyl)ated by PARP. PARP has also been shown to activate DNA-dependent protein kinase (DNA-PK) activity in vitro and thus to regulate the activity of p53 by phosphorylation (Ruscetti et al. 1998). The PARP inhibitor 3AB also acts as an inhibitor of tumour promotion in mouse skin (Ludwig et al. 1990). On the other hand, it can act as a cocarcinogen for UV-induced carcinogenesis (Epstein & Cleaver 1992). Altogether, the relationship between PARP and p53 seems to differ in different models and still needs further studies to be thoroughly understood.
2.4.4.2. Oncogenic Ras
Mammalian ras genes are considered crucial in the regulation of cell proliferation (Johnson et al. 1997, reviewed by Bos 1989). In mammals, the ras family consists of three genes located in different chromosomes, encoding the homologous 21 kDa proteins H-Ras, N-Ras and K-Ras. It has been estimated that 30% of all human cancers express mutated forms of ras (for a review, see McMahon & Woods 2001). The signal of Ras can have either negative or positive effects on cell growth, differentiation and death (reviewed by Frame & Balmain 2000). The signal is subsequently transmitted by a cascade of kinases, which results in the activation of MAPK. The Ras-MAPK pathway is apparently involved in the regulation of basal and induced levels of p53 (Fukasawa & Vande Woude 1997, Serrano et al. 1997). In vascular smooth muscle cells, BP treatment has been shown to cause an increase in Ras mRNA levels (Kerzee & Ramos 2000). Ras, in turn, induces p19ARF in murine fibroblasts (Ferbeyre et al. 2002, Groth et al. 2000). There are also data that support a linear model from Ras through the induction of p19ARF to p53. Palmero and coworkers (1998) showed that an oncogenic form of Ras protein increases significantly p19ARF mRNA. Also, in ARF-/- mouse embryonic fibroblasts (MEF), the p53 level is not affected by oncogenic Ras. In an earlier work on wild-type MEFs, the p53 level increased after oncogenic Ras (Serrano et al. 1997). It can thus be concluded that p19ARF is required for oncogenic Ras-induced accumulation of p53 (Fig. 3).
2.4.4.3. p21WAF1/Cip1/Sdi1
The p21 protein was the first cyclin-dependent kinase (CDK) identified (El-Deiry et al. 1993, Harper et al. 1993, Noda et al. 1994). The p21 protein has multiple functions. El-Deiry and coworkers (1993) named this gene WAF1 and found it to code a protein that mediates p53-induced growth arrest of the cell cycle. Almost simultaneously, another group showed it to be a regulator of CDK activity (Harper et al. 1993). Yet another group demonstrated its gene expression to be induced in relation to cellular senescence (Noda et al. 1994). p21 can inhibit CDK-cyclin activity (reviewed by Boulaire et al. 2000) and directly inhibit DNA replication (Li et al. 1994, Shivji et al. 1994, Chen et al. 1995). The gene is transcriptionally upregulated by wild-type p53 (El-Deiry et al. 1993). The activation of p53 causes induction of p21, which in turn inhibits CDK-cyclin activity and arrests the cell cycle at the G1 (reviewed by Gartel et al. 1996, Colman et al. 2000) or G2 (reviewed by Taylor & Stark 2001, Winters 2002) cell cycle checkpoint. This gives time for DNA repair before replication or mitosis and thus links p21 directly to the tumour suppressor function of p53.
2.4.4.4. “Family members” of p53: p63 and p73 proteins
Recently, two new genes notably similar to the TP53 gene have been found. One of these genes is called p63, p51 or KET, (Yang et al. 1998a, Osada et al. 1998, Schmale & Bamberger 1997, respectively) and the other p73 (Kaghad et al. 1997). They encode proteins that share high sequence similarity and conserved functional domains with p53 and can exert p53-like functions, such as transactivation of p53 target genes and induction of apoptosis (reviewed by Yang et al. 2002). Both give rise to differentially spliced mRNAs and, respectively, to several different proteins homologous to p53 (reviewed by Levrero et al. 2000). There are at least three different forms of the p63 protein differing at the C-terminal end (α, β and γ ) that may also differ at the transactivation domain (p63TA and p63ΔDN) and six different variants of the p73 protein, p73-. The p73 protein, like p53, accumulates in response to DNA damage, and it is noteworthy that different types of inducers of DNA damage seem to affect p73 in different ways (reviewed by Levrero et al. 2000). Both p63 and p73 take part in the regulation of normal cell development and apoptosis (reviewed by Lohrum & Vousden 2000). Different forms of p63 protein can act in a dominant-negative manner towards p53 (Yang et al. 1998a), but whether p63 dysregulation has a role in tumorigenesis remains to be seen. p73, on the other hand, has been suggested to be a tumour suppressor protein (see Levrero et al. 2000), although opposite opinions have also been presented (Irwin & Kaelin 2001). The function of p63 or p73 as a tumour suppressor still remains unclear (reviewed by Michael & Oren 2002).
2.4.5. p53 in vitro and in vivo
Because of its importance in human cancers, p53 is nowadays one of the most intensively studied proteins. Significant progress has been made in the recent years, including the identification of the types of cellular stress that induce p53, identification of the post-translational modifications that regulate p53 and characterization of the proteins that interact with p53 and regulate its function (see Fig. 3). All of these important findings have been made in vitro. The question that still remains to be answered concerns the relevance of these results in vivo (reviewed by Sansom & Clarke 2000). The significance of individual aspects concerning the p53 protein, including its function, modification and regulation, needs to be studied in vivo. The progress in understanding the p53 pathway in vitro and the introduction of transgenic mice models (reviewed by Herzig & Christofori 2002) will facilitate this work.